MHC binding prediction based on modeled physicochemical properties of peptides

Project description
MHCLovac
MHC binding prediction based on modeled physicochemical properties of peptides
New in version 4.0
- Training data with 4+ million data samples, binding affinity + eluted ligands.
- Separate ligand prediction from the previous version is removed.
- Binding score is reported as the log transformed binding affinity:
1 - log50k(affinity).
Table of content
About
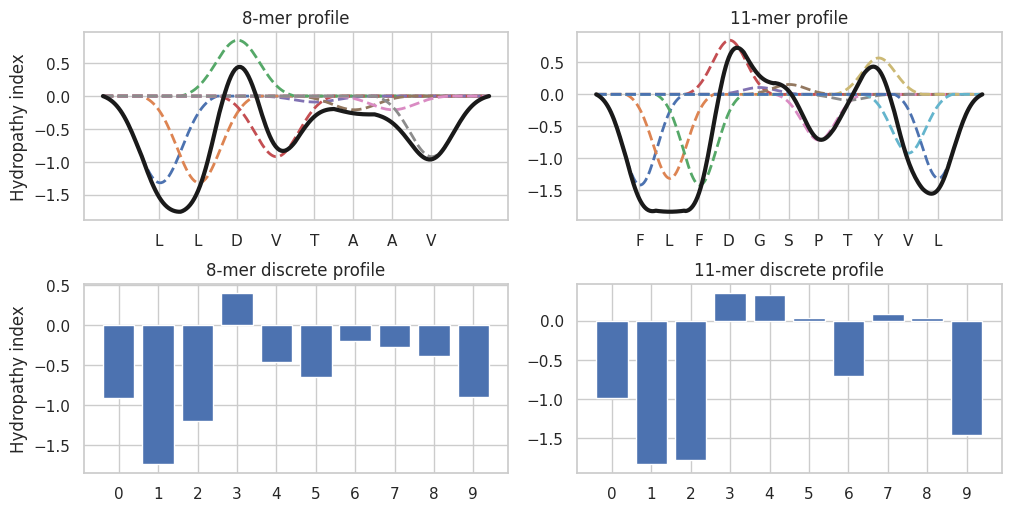
MHCLovac is MHC binding prediction method that focuses on physicochemical properties of peptides responsible for interaction with MHC molecules. This method is based on modeling physicochemical properties of peptides in a way that captures the nearest neighbor effect of amino acid residues. In other words, the method is based on the following assumption: physicochemical properties of adjacent amino acid residues have additive effect on the local properties of the peptide as a whole, and properties of a single residue affect the properties of the peptide at the neighboring positions. Using this approach each peptide is represented by a set of modeled physicochemical profiles (distributions of certain property, figure 1, upper subplots) which are further reduced to predetermined number of discrete data points to obtain discrete physicochemical profiles (figure 1, bottom subplots). Discrete profiles are used as input features for binding prediction models. This method allows for direct comparison of physicochemical profiles of peptides of different sequence lengths.
Materials
Training data was obtained from NetMHCPan 4.1 (Reynisson, B. et. al., 2020) website and contains preprocessed binding affinity and eluted ligand data. In addition, binding affinity data was obtained directly from IEDB (Vita R et. al., 2018), and this dataset was used to narrow down the set of physicochemical indexes which are used for prediction. The list of physicochemical properties and corresponding amino acid index data was obtained from the AAindex database (Kawashima, S. et. al, 2008).
Methods
MHCLovac uses a collection of out-of-the-box regression algorithms from scikit-learn python library with mostly default parameters.
The prediction model returns binding scores in form of log transformed binding affinity (1 – log50k(affinity)).
Input features for prediction models are discrete physicochemical profiles of peptides.
Since the AAindex database contains more than 500 entries, to reduce the number of physicochemical properties needed to model, the following selection method is implemented:
for each physicochemical property index, the binding model was trained and evaluated using r2 score for each MHC allele, and the average score across all alleles was calculated.
The indexes were sorted based on the average score in descending order.
Starting from the highest scoring index (selected by default), each next index was compared to the previously selected ones for correlation coefficient.
Only if correlation coefficients with all indexes from selection were in range [-0.3, 0.3] the new index was added to the selection.
This resulted in total of 9 indexes (table 1) which had high prediction potential for most alleles and were also low-correlated between themselves.
| Accession number | Title | Average r2 score |
|---|---|---|
| ROSM880102 | Side chain hydropathy, corrected for solvation (Roseman, 1988) | 0.2819 |
| ZIMJ680104 | Isoelectric point (Zimmerman et al., 1968) | 0.2740 |
| OOBM770104 | Average non-bonded energy per residue (Oobatake-Ooi, 1977) | 0.2498 |
| SNEP660101 | Principal component I (Sneath, 1966) | 0.2454 |
| ROBB760111 | Information measure for C-terminal turn (Robson-Suzuki, 1976) | 0.2353 |
| CHAM830102 | A parameter defined from the residuals obtained from the best correlation of the Chou-Fasman parameter of beta-sheet (Charton-Charton, 1983) | 0.2062 |
| RACS820104 | Average relative fractional occurrence in EL(i) (Rackovsky-Scheraga, 1982) | 0.1833 |
| WERD780103 | Free energy change of alpha(Ri) to alpha(Rh) (Wertz-Scheraga, 1978) | 0.1704 |
| KARS160120 | Weighted minimum eigenvalue based on the atomic numbers (Karkbara-Knisley, 2016) | 0.1413 |
Results
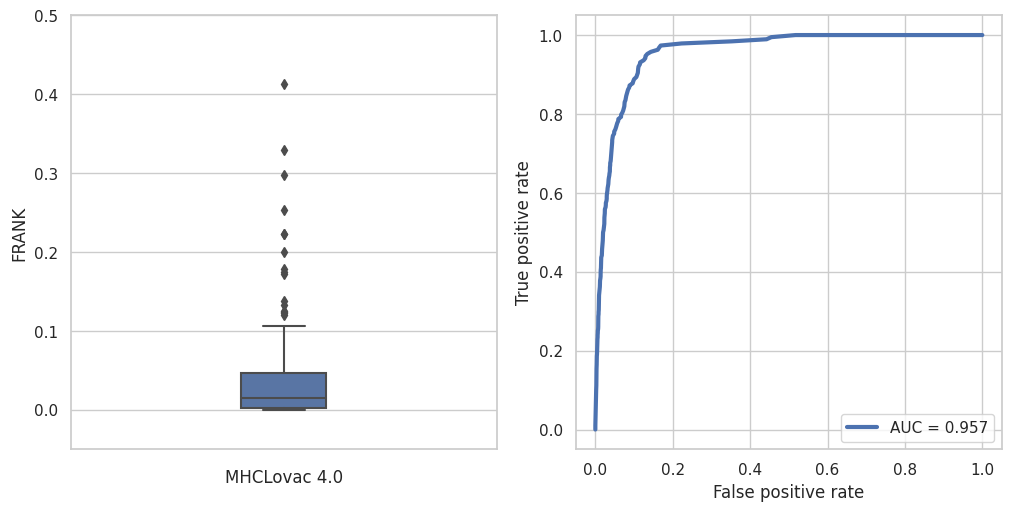
Prediction is evaluated using FRANK method from NetMHCPan 4.1 paper. Dataset was also obtained from NetMHCPan website and contains some 1600 sequences and corresponding epitopes of which 200 were randomly selected for this benchmark. FRANK score measures the fraction of non-epitopes scoring higher than the epitope (from same sequence), and the best possible score is 0.
Installation
pip install mhclovac
Usage
From command line:
mhclovac -f example.fasta -m HLA-B*44:02 -l 11 --sort --n_cpu 6
Programmatically:
from mhclovac import predict
from mhclovac.utils import list_mhc_alleles
alleles = list_mhc_alleles()
# returns list of supported MHC alleles
predictions = predict(
peptides=['MEIFIEVFSHF', 'LELPTGSLEKS', 'TELTLNMCLEL'],
mhc_allele='HLA-B*44:02',
sort=True,
n_cpu=6
)
# returns pandas DataFrame with prediction results
Example output:
peptide mhc_allele peptide_length sequence_name binding_score
MEIFIEVFSHF HLA-B*44:02 11 MEIFIEVFSHF HLA-B44:02 0.626139
LELPTGSLEKS HLA-B*44:02 11 MEIFIEVFSHF HLA-B44:02 0.211701
TELTLNMCLEL HLA-B*44:02 11 MEIFIEVFSHF HLA-B44:02 0.185610
IEVFSHFLLQL HLA-B*44:02 11 MEIFIEVFSHF HLA-B44:02 0.171749
LEKSLMISSQV HLA-B*44:02 11 MEIFIEVFSHF HLA-B44:02 0.147054
References
- Reynisson, B., Alvarez, B., Paul, S., Peters, B., & Nielsen, M. (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Research. https://doi.org/10.1093/nar/gkaa379
- Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2018 Oct 24. doi: 10.1093/nar/gky1006. [Epub ahead of print] PubMed PMID: 30357391. https://doi.org/10.1093/nar/gky1006
- Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T., and Kanehisa, M.; AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202-D205 (2008). [PMID:17998252] https://doi.org/10.1093/nar/gkm998


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.







