A multi-sample fastqc pipeline from Sequana project

Project description




This is is the fastqc pipeline from the Sequana projet
- Overview:
Runs fastqc and multiqc on a set of Sequencing data to produce control quality reports
- Input:
A set of FastQ files (paired or single-end) compressed or not
- Output:
An HTML file summary.html (individual fastqc reports, mutli-samples report)
- Status:
Production
- Wiki:
- Documentation:
This README file, the Wiki from the github repository (link above) and https://sequana.readthedocs.io
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352
Installation
sequana_fastqc is based on Python3, just install the package as follows:
pip install sequana_fastqc --upgrade
You will need third-party software such as fastqc. Please see below for details.
Usage
If you have a set of FastQ files in a data/ directory, type:
sequana_fastqc --input-directory data
To know more about the options (e.g., add a different pattern to restrict the execution to a subset of the input files, change the output/working directory, etc):
sequana_fastqc --help
The call to sequana_fastqc creates a directory fastqc. Then, you go to the working directory and execute the pipeline as follows:
cd fastqc sh fastqc.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the fastqc.rules and config.yaml files and then execute the pipeline yourself with specific parameters:
snakemake -s fastqc.rules --cores 4 --stats stats.txt
Or use sequanix interface.
Please see the Wiki for more examples and features.
Tutorial
You can retrieve test data from sequana_fastqc (https://github.com/sequana/fastqc) or type:
wget https://raw.githubusercontent.com/sequana/fastqc/master/sequana_pipelines/fastqc/data/data_R1_001.fastq.gz wget https://raw.githubusercontent.com/sequana/fastqc/master/sequana_pipelines/fastqc/data/data_R2_001.fastq.gz
then, prepare the pipeline:
sequana_fastqc --input-directory . cd fastqc sh fastq.sh # once done, remove temporary files (snakemake and others) make clean
Just open the HTML entry called summary.html. A multiqc report is also available. You will get expected images such as the following one:
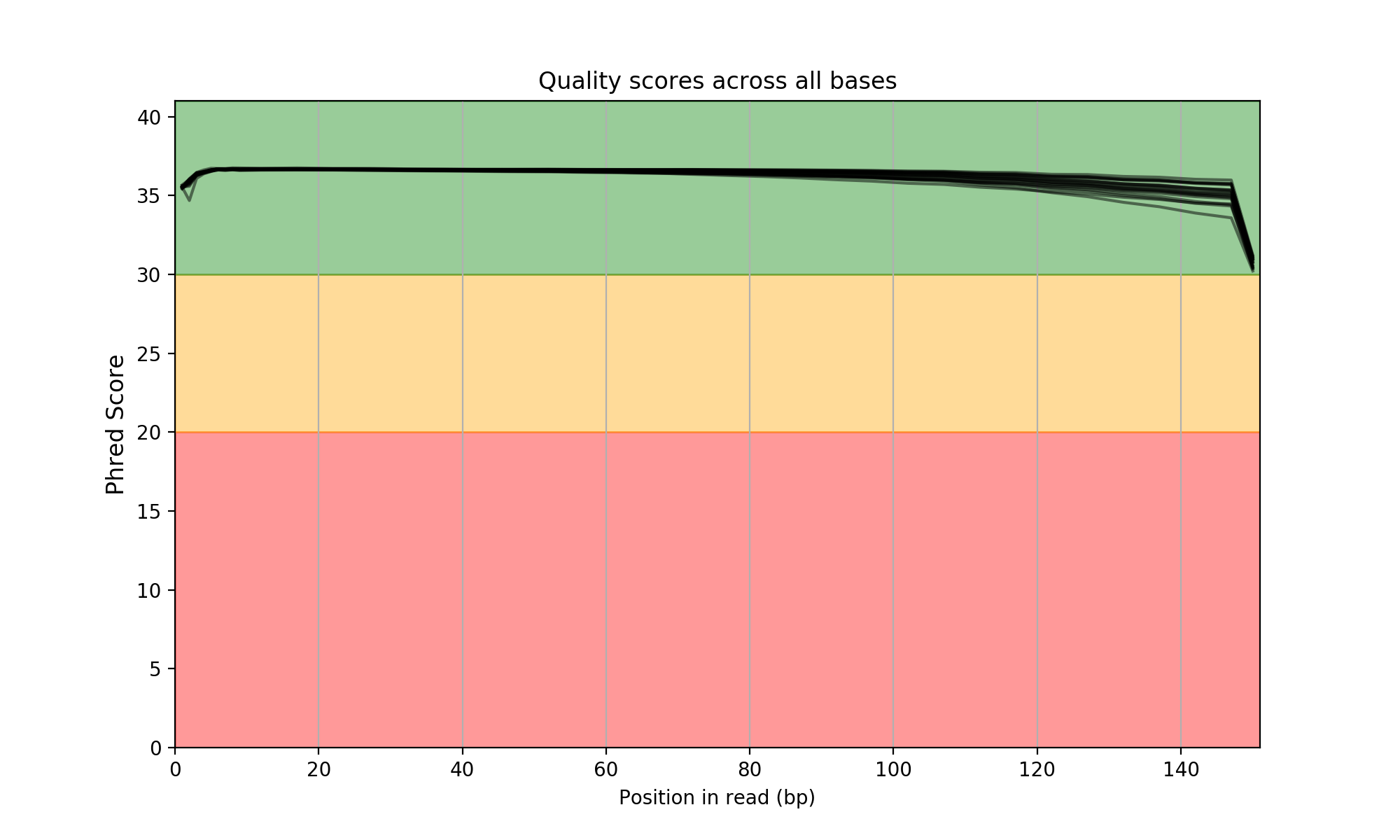
Please see the Wiki for more examples and features.
Requirements
This pipelines requires the following executable(s):
fastqc
falco (optional)
For Linux users, we provide apptainer/singularity images available through the damona project (https://damona.readthedocs.io).
To make use of them, initiliase the pipeline with the –use-apptainer option and everything should be downloaded automatically for you, which also guarantees reproducibility:
sequana_fastqc --input-directory data --use-apptainer --apptainer-prefix ~/images
Details
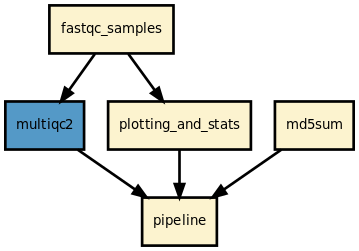
This pipeline runs fastqc in parallel on the input fastq files (paired or not) and then execute multiqc. A brief sequana summary report is also produced. s You may use falco instead of fastqc. This is experimental but seem to work for Illumina/FastQ files.
This pipeline has been tested on several hundreds of MiSeq, NextSeq, MiniSeq, ISeq100, Pacbio runs.
It produces a md5sum of your data. It copes with empty samples. Produces ready-to-use HTML reports, etc
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Changelog
Version |
Description |
|---|---|
1.8.2 |
|
1.8.1 |
|
1.8.0 |
|
1.7.1 |
|
1.7.0 |
|
1.6.2 |
|
1.6.1 |
|
1.6.0 |
|
1.5.0 |
|
1.4.2 |
|
1.4.1 |
|
1.4.0 |
|
1.3.0 |
|
1.2.0 |
|
1.1.0 |
|
1.0.1 |
|
1.0.0 |
|
0.9.15 |
|
0.9.14 |
|
0.9.13 |
|
0.9.12 |
|
0.9.11 |
|
0.9.10 |
|
0.9.9 |
|
0.9.8 |
|
0.9.7 |
|
0.9.6 |
add the readtag option |
Contribute & Code of Conduct
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Hashes for sequana_fastqc-1.8.2-py3-none-any.whl
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | 6cd0e380870833791bb75b426a96e14e75bf447ae7eaba1f72b2cd3cfdb2f1b9 |
|
| MD5 | 46c20a7f880e7e1b021257beaa70652a |
|
| BLAKE2b-256 | 100e94eaa0e13b9994c567f513fcad632d94f979c8d38587d8792c9e8a7ad26b |







