Assess the quality of metagenome-assembled viral genomes.


Project description

CheckV is a fully automated command-line pipeline for assessing the quality of metagenome-assembled viral genomes, including identification of host contamination for integrated proviruses, estimating completeness for genome fragments, and identification of closed genomes.
The pipeline can be broken down into 4 main steps:
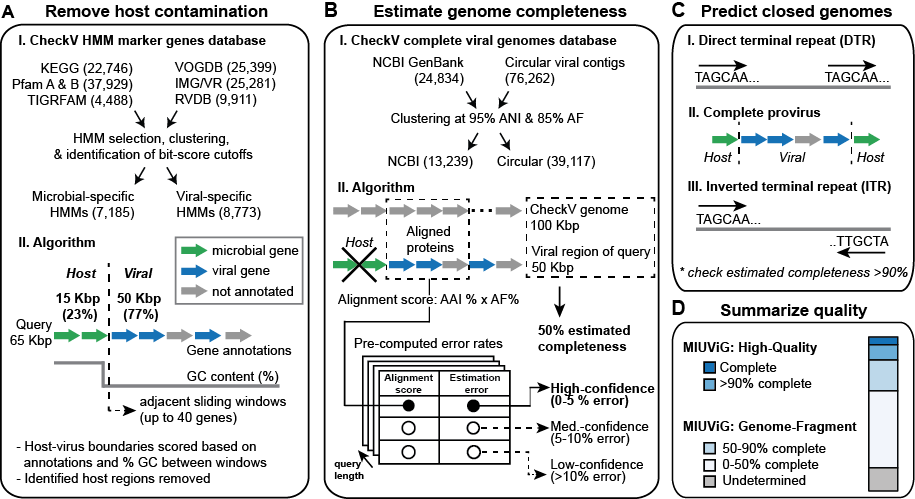
A: Remove host contamination. CheckV identifies and removes non-viral regions on proviruses. Genes are first annotated based on comparison to a custom database of HMMs that are highly specific to either viral or microbial proteins. Next, the program compares the gene annotations and GC content between a pair of sliding windows that each contain up to 40 genes. This information is used to compute a score at each intergenic position and identify host-virus boundaries.
B: Estimate genome completeness. CheckV estimates genome completeness in two stages. First, proteins are compared to the CheckV genome database using AAI (average amino acid identity), completeness is computed as a simple ratio between the contig length (or viral region length for proviruses) and the length of matched reference genomes, and a confidence level is reported. In some cases, a contig won't have a high- or medium-confidence estimate based on AAI. In these cases, a more sensitive but less accurate approach is used based on HMMs shared between the contig and CheckV reference genomes (ANI: average nucleotide identity; AF: alignment fraction)
C: Predict closed genomes. Closed genomes are identified based either on direct terminal repeats (DTRs; often indicating a circular sequence), flanking virus-host boundaries (indicating a complete prophage), or inverted terminal repeats (ITRs; believed to facilitate circularization and recombination). Whenever possible, these predictions are validated based on the estimated completeness obtained in B (e.g. completeness >90%). DTRs are the most reliable and most common indicator of complete genomes.
D: Summarize quality. Based on the results of A-C, CheckV generates a report file and assigns query contigs to one of five quality tiers: complete, high-quality (>90% completeness), medium-quality (50-90% completeness), low-quality (<50% completeness), or undetermined quality.
Installation
There are two methods to install CheckV:
- Using
conda:
conda install -c conda-forge -c bioconda checkv
- Using
pip:
pip install checkv
If you decide to install CheckV via pip, make sure you also have the following external dependencies installed:
- BLAST+ (v2.5.0)
- DIAMOND (v0.9.30)
- HMMER (v3.3)
- Prodigal (v2.6.3)
The versions listed above were the ones that were properly tested. Different versions may also work.
CheckV database
Whichever method you choose to install CheckV you will need to download and extract database in order to use it:
wget https://portal.nersc.gov/CheckV/checkv-db-v0.6.tar.gz
tar -zxvf checkv-db-v0.6.tar.gz
Update your environment:
export CHECKVDB=/path/to/checkv-db-v0.6
If you don't want to set the environmet variable, you can still use the database through the -d parameter of the contamination and completeness modules.
Quick start
Navigate to CheckV test directory:
cd /path/to/checkv/test
Identify flanking host regions on integrated prophages:
checkv contamination test.fna checkv_out -t 16
Estimate completeness for genome fragments:
checkv completeness test.fna checkv_out -t 16
Identify (possible) complete genomes with terminal: (this module also estimates the genome copy number; see below for details)
checkv repeats test.fna checkv_out
Summarize CheckV output & classify contigs into quality tiers:
checkv quality_summary test.fna checkv_out
For optimal results, you should always run the 4 steps in this order.
Frequently asked questions
Q: What is the difference between AAI- and HMM-based completeness?
A: AAI-based completeness was designed to be very accurate and can be trusted when the confidence is medium or high. HMM-based completeness was designed to confidently estimate the minimum completeness. So a value of 50% indicates that we can be 95% sure that the viral contig is at least 50% complete. But it may be more complete, so this should be taken into consideration when analyzing CheckV output.
Q: What is the meaning of the genome_copies field?
A: This is a measure of how many times the viral genome is represented in the contig. Most times this is 1.0 (or very close to 1.0). In rare cases assembly errors may occur in which the contig sequence represents multiple concatenated copies of the viral genome. In these cases genome_copies will exceed 1.0.
Q: Why does my DTR contig have <100% estimated completeness? A: If the estimated completeness is close to 100% (e.g. 90-110%) then the query is likely complete. However sometimes incomplete genome fragments may contain a direct terminal repeat (DTR), in which case we should expect their estimated completeness to be <90%, and sometimes much less. In other cases, the contig will truly be circular, but the estimated completeness is incorrect. This may also happen if the query a complete segment of a multipartite genome (common for RNA viruses). By default, CheckV uses the 90% completeness cutoff for verification, but a user may wish to make their own judgement in these ambiguous cases.
Q: Why is my DTR contig predicted as a provirus? A: CheckV classifies a sequence as a provirus if it is contains a host region (usually occuring on one just side of the sequence). A DTR sequence represents a complete viral genome, so these predictions are at odds with eachother and indicate either a false positive DTR prediction, or a false positive provirus prediction. By default, CheckV considers these complete genomes, but a user may wish to make their own judgement in these ambiguous cases.
Q: Why is my sequence considered "high-quality" when it has high contamination? A: CheckV determines sequence quality solely based on completeness. Host contamination is easily removed, so is not factored into these quality tiers.
Q: I performed binning and generated viral MAGs. Can I use CheckV on these? A: CheckV can estimate completeness but not contamination for these. Additionally, you'll need to concatentate the contigs from each MAG into a single sequence prior to running CheckV.
Q: Can I apply CheckV to eukaryotic viruses? A: Probably, but this has not been tested. The reference database includes a large number of genomes and HMMs that should match eukaryotic genomes. However, CheckV may report a completeness <90% if your genome is a single segment of a segmented viral genome. CheckV may also classify your sequence as a provirus if it contains a large island of metabolic genes commonly found in bacteria/archaea.
Q: Can I use CheckV to predict (pro)viruses from whole (meta)genomes? A: Possibly, though this has not been tested.
Q: How should I handle putative "closed genomes" with no completeless estimate? A: In some cases, you won't be able to verify the completeness of a sequence with terminal repeats or provirus integration sites. DTRs are a fairly reliable indicator (>90% of the time) and can likely be trusted with no completeness estimate. However, complete proviruses and ITRs are much less reliable indicators, and therefore require >90% estimated completeness.
Q: Why is my contig classified as "undetermined quality"? A: This happens when the sequence doesn't match any CheckV reference genome with high enough similarity to confidently estimate completeness. There are a few explanations for this, in order of likely frequency: 1) your contig is very short, and by chance it does not share any genes with a CheckV reference, 2) your contig is from a very novel virus that is distantly related to all genomes in the CheckV database, 3) your contig is not a virus at all and so doesn't match any of the references.
Q: How should I handle sequences with "undetermined quality"? A: While it is not possible to estimate completeness for these, you may choose to still analyze sequences above a certain length (e.g. >30 kb). If you have knowledge about the viral clade, then this information can be taken into account (e.g. keep >5 kb sequences from Microviridae). Or you can use these sequences in analyses that don't require high-quality genomes.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.







