A genome visualization python package for comparative genomics

Project description
pyGenomeViz






Overview
pyGenomeViz is a genome visualization python package for comparative genomics implemented in matplotlib. This package is developed for the purpose of easily and beautifully plotting genomic features and sequence similarity comparison links between multiple genomes. It supports genome visualization of Genbank format file and can be saved figure in various formats (JPG/PNG/SVG/PDF). User can use pyGenomeViz for interactive genome visualization figure plotting on jupyter notebook, or automatic genome visualization figure plotting in genome analysis scripts/pipelines.
For more information, please see full documentation here.
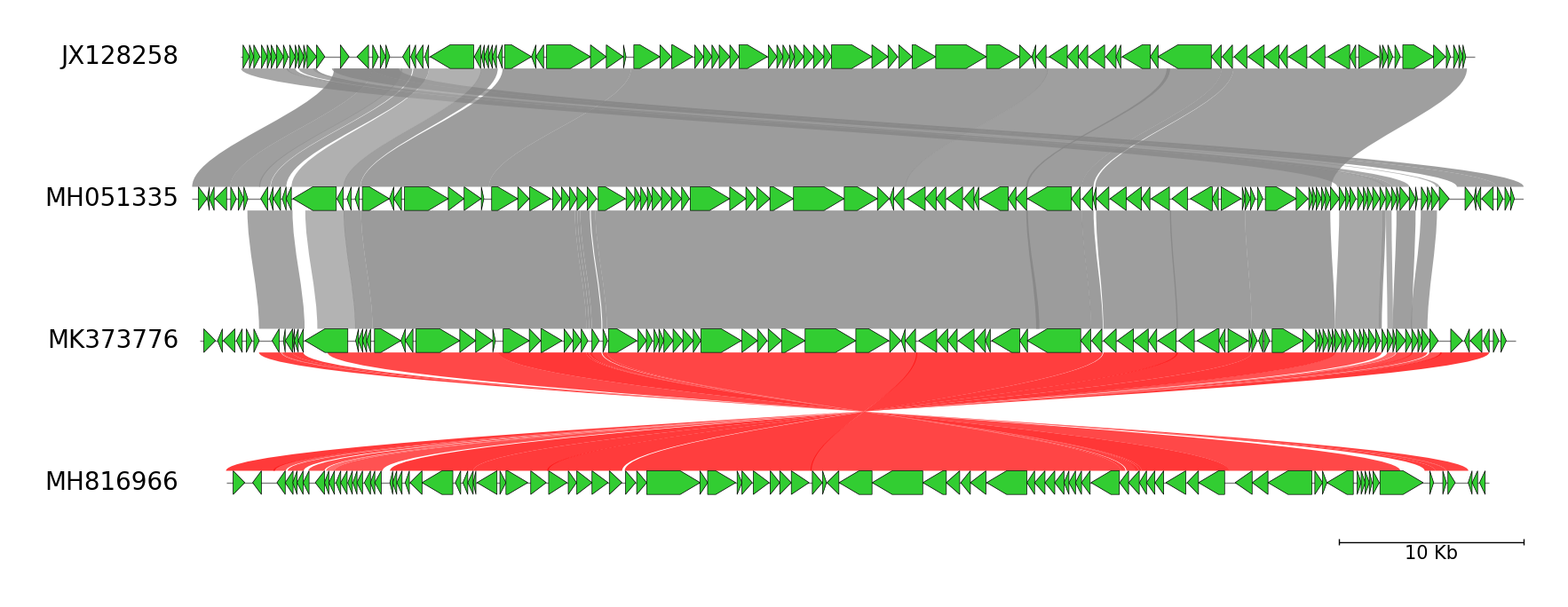
Fig.1 Four Erwinia phage genome comparison result
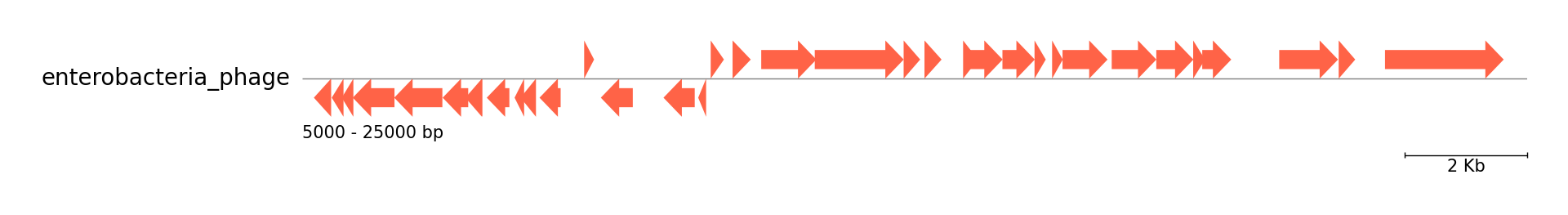
Fig.2 Six Enterobacteria phage genome comparison result
Installation
Python 3.7 or later is required for installation.
Install PyPI package:
pip install pygenomeviz
Install bioconda package:
conda install -c conda-forge -c bioconda pygenomeviz
Examples
Jupyter notebooks containing code examples below is available here.
Basic Example
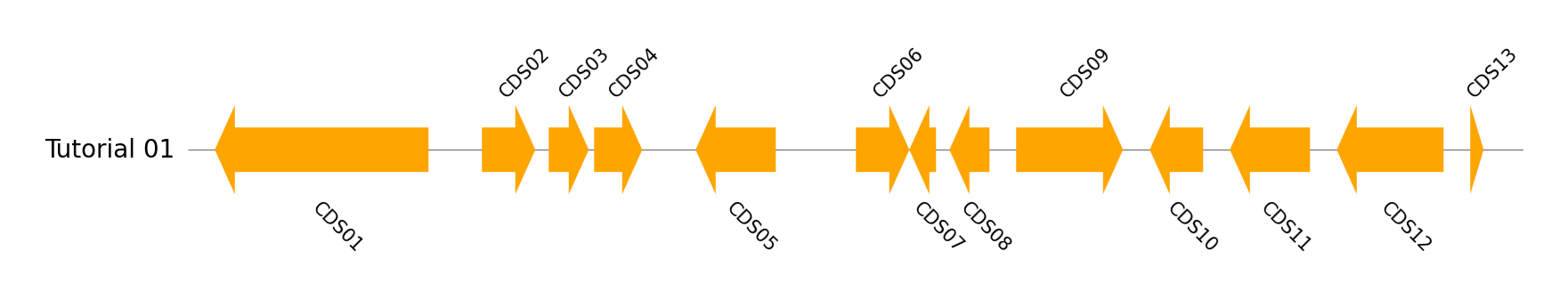
Single Genome Track
from pygenomeviz import GenomeViz
name, genome_size = "Tutorial 01", 5000
cds_list = ((100, 900, -1), (1100, 1300, 1), (1350, 1500, 1), (1520, 1700, 1), (1900, 2200, -1), (2500, 2700, 1), (2700, 2800, -1), (2850, 3000, -1), (3100, 3500, 1), (3600, 3800, -1), (3900, 4200, -1), (4300, 4700, -1), (4800, 4850, 1))
gv = GenomeViz()
track = gv.add_feature_track(name, genome_size)
for idx, cds in enumerate(cds_list, 1):
start, end, strand = cds
track.add_feature(start, end, strand, label=f"CDS{idx:02d}")
gv.savefig("example01.png")
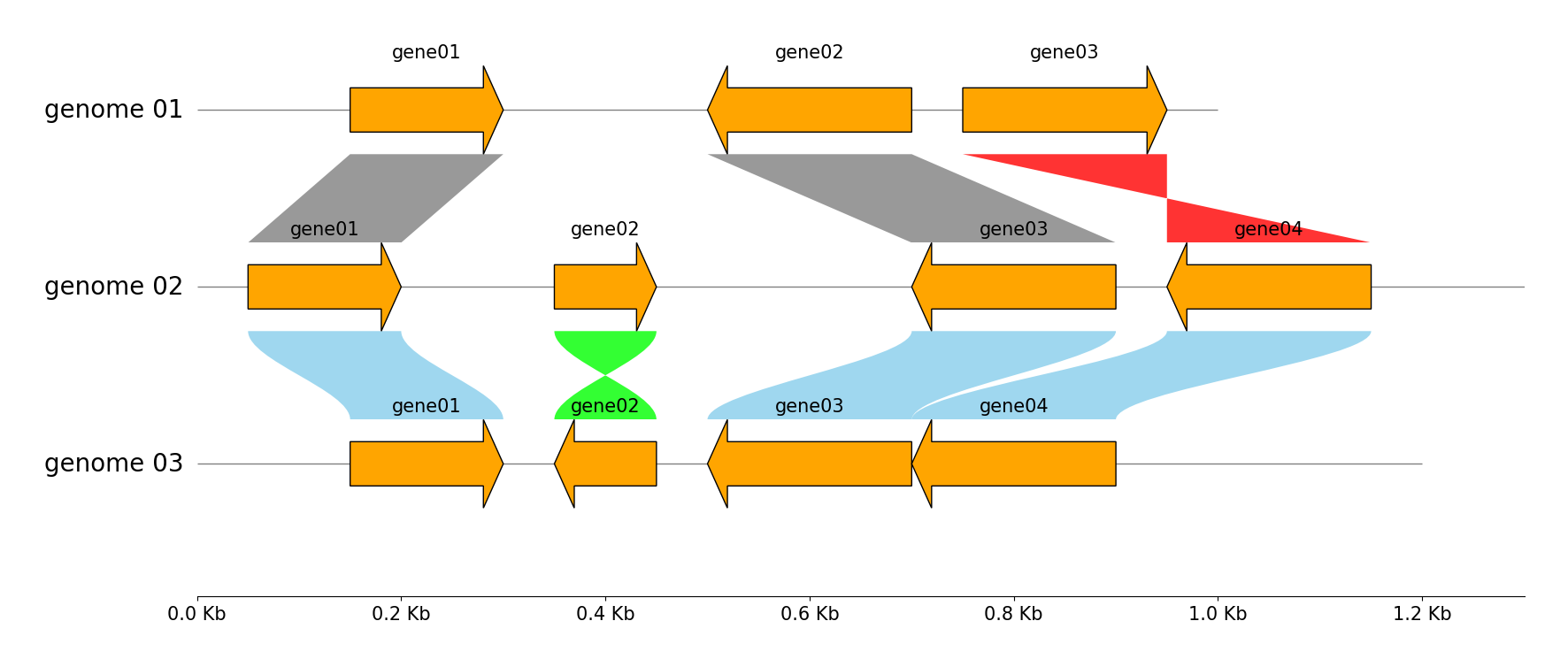
Multiple Genome Tracks & Links
from pygenomeviz import GenomeViz
genome_list = (
{"name": "genome 01", "size": 1000, "cds_list": ((150, 300, 1), (500, 700, -1), (750, 950, 1))},
{"name": "genome 02", "size": 1300, "cds_list": ((50, 200, 1), (350, 450, 1), (700, 900, -1), (950, 1150, -1))},
{"name": "genome 03", "size": 1200, "cds_list": ((150, 300, 1), (350, 450, -1), (500, 700, -1), (701, 900, -1))},
)
gv = GenomeViz(tick_style="axis")
for genome in genome_list:
name, size, cds_list = genome["name"], genome["size"], genome["cds_list"]
track = gv.add_feature_track(name, size)
for idx, cds in enumerate(cds_list, 1):
start, end, strand = cds
track.add_feature(start, end, strand, label=f"gene{idx:02d}", linewidth=1, labelrotation=0, labelvpos="top", labelhpos="center", labelha="center")
# Add links between "genome 01" and "genome 02"
gv.add_link(("genome 01", 150, 300), ("genome 02", 50, 200))
gv.add_link(("genome 01", 700, 500), ("genome 02", 900, 700))
gv.add_link(("genome 01", 750, 950), ("genome 02", 1150, 950))
# Add links between "genome 02" and "genome 03"
gv.add_link(("genome 02", 50, 200), ("genome 03", 150, 300), normal_color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 350, 450), ("genome 03", 450, 350), normal_color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 900, 700), ("genome 03", 700, 500), normal_color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 03", 900, 701), ("genome 02", 1150, 950), normal_color="skyblue", inverted_color="lime", curve=True)
gv.savefig("example02.png")
Practical Example
Single Genome Track from Genbank file
from pygenomeviz import Genbank, GenomeViz, load_dataset
gbk_files, _ = load_dataset("enterobacteria_phage")
gbk = Genbank(gbk_files[0])
gv = GenomeViz()
track = gv.add_feature_track(gbk.name, gbk.genome_length)
track.add_genbank_features(gbk)
gv.savefig("example03.png")
Multiple Genome Tracks & Links from Genbank files
from pygenomeviz import Genbank, GenomeViz, load_dataset
gv = GenomeViz(
fig_track_height=0.7,
feature_track_ratio=0.2,
tick_track_ratio=0.7,
tick_style="bar",
align_type="center",
)
gbk_files, links = load_dataset("escherichia_phage")
for gbk_file in gbk_files:
gbk = Genbank(gbk_file)
track = gv.add_feature_track(gbk.name, gbk.genome_length)
track.add_genbank_features(gbk, facecolor="limegreen", linewidth=0.5, arrow_shaft_ratio=1.0)
for link in links:
link_data1 = (link.ref_name, link.ref_start, link.ref_end)
link_data2 = (link.query_name, link.query_start, link.query_end)
gv.add_link(link_data1, link_data2, v=link.identity, curve=True)
gv.savefig("example04.png")

Customization Tips
Since pyGenomeViz is implemented based on matplotlib, users can easily customize the figure in the manner of matplotlib. Here are some tips for figure customization.
Customization Tips 01
- Add
GC Content&GC skewsubtrack - Add annotation label & fillbox
- Add colorbar for links identity
Code
from pygenomeviz import Genbank, GenomeViz, load_dataset
gv = GenomeViz(
fig_width=12,
fig_track_height=0.7,
feature_track_ratio=0.5,
tick_track_ratio=0.3,
tick_style="axis",
)
gbk_files, links = load_dataset("erwinia_phage")
gbk_list = [Genbank(gbk_file) for gbk_file in gbk_files]
for gbk in gbk_list:
track = gv.add_feature_track(gbk.name, gbk.genome_length)
track.add_genbank_features(gbk, plotstyle="arrow")
min_identity = int(min(link.identity for link in links))
for link in links:
link_data1 = (link.ref_name, link.ref_start, link.ref_end)
link_data2 = (link.query_name, link.query_start, link.query_end)
gv.add_link(link_data1, link_data2, v=link.identity, vmin=min_identity)
# Add subtracks to top track for plotting 'GC content' & 'GC skew'
gv.add_feature_subtrack(gv.top_track.name, "gc_content", ratio=0.7)
gv.add_feature_subtrack(gv.top_track.name, "gc_skew", ratio=0.7)
fig = gv.plotfig()
# Add label annotation to top track
top_track = gv.top_track # or, gv.get_track("MT939486") or gv.get_tracks()[0]
label, start, end = "Inverted", 310000 + top_track.offset, 358000 + top_track.offset
center = int((start + end) / 2)
top_track.ax.hlines(1.5, start, end, colors="red", linewidth=1, linestyles="dashed", clip_on=False)
top_track.ax.text(center, 2.0, label, fontsize=12, color="red", ha="center", va="bottom")
# Add fillbox to top track
x, y = (start, start, end, end), (1, -1, -1, 1)
top_track.ax.fill(x, y, fc="lime", linewidth=0, alpha=0.1, zorder=-10)
# Plot GC content for top track
gc_content_ax = gv.top_track.subtracks[0].ax # or, gv.get_track("gc_content").ax
pos_list, gc_content_list = gbk_list[0].calc_gc_content()
gc_content_ax.set_ylim(bottom=0, top=max(gc_content_list))
pos_list += gv.top_track.offset # Offset is required if align_type is not 'left'
gc_content_ax.fill_between(pos_list, gc_content_list, alpha=0.2, color="blue")
gc_content_ax.text(gv.top_track.offset, max(gc_content_list) / 2, "GC(%) ", ha="right", va="center", color="blue")
# Plot GC skew for top track
gc_skew_ax = gv.top_track.subtracks[1].ax # or, gv.get_track("gc_skew").ax
pos_list, gc_skew_list = gbk_list[0].calc_gc_skew()
gc_skew_abs_max = max(abs(gc_skew_list))
gc_skew_ax.set_ylim(bottom=-gc_skew_abs_max, top=gc_skew_abs_max)
pos_list += gv.top_track.offset # Offset is required if align_type is not 'left'
gc_skew_ax.fill_between(pos_list, gc_skew_list, alpha=0.2, color="red")
gc_skew_ax.text(gv.top_track.offset, 0, "GC skew ", ha="right", va="center", color="red")
# Set coloarbar for link
gv.set_colorbar(fig, vmin=min_identity)
fig.savefig("example05.png", bbox_inches="tight")
Customization Tips 02
- Add legends
- Add colorbar for links identity
Code
from matplotlib.lines import Line2D
from matplotlib.patches import Patch
from pygenomeviz import Genbank, GenomeViz, load_dataset
gv = GenomeViz(
fig_width=10,
fig_track_height=0.7,
feature_track_ratio=0.5,
tick_track_ratio=0.5,
align_type="center",
tick_style="bar",
tick_labelsize=10,
)
gbk_files, links = load_dataset("enterobacteria_phage")
for idx, gbk_file in enumerate(gbk_files):
gbk = Genbank(gbk_file)
track = gv.add_feature_track(gbk.name, gbk.genome_length, labelsize=10)
track.add_genbank_features(
gbk,
label_type="product" if idx == 0 else None, # Labeling only top track
label_filter=["hypothetical"], # Ignore 'hypothetical ~~~' label
labelsize=8,
labelvpos="top",
facecolor="skyblue",
linewidth=0.5,
)
normal_color, inverted_color, alpha = "chocolate", "limegreen", 0.5
min_identity = int(min(link.identity for link in links))
for link in links:
link_data1 = (link.ref_name, link.ref_start, link.ref_end)
link_data2 = (link.query_name, link.query_start, link.query_end)
gv.add_link(link_data1, link_data2, normal_color, inverted_color, alpha, v=link.identity, vmin=min_identity, curve=True)
fig = gv.plotfig()
# Add Legends (Maybe there is a better way)
handles = [
Line2D([], [], marker=">", color="skyblue", label="CDS", ms=10, ls="none"),
Patch(color=normal_color, label="Normal Link"),
Patch(color=inverted_color, label="Inverted Link"),
]
fig.legend(handles=handles, frameon=True, bbox_to_anchor=(1, 0.8), loc="upper left", ncol=1, handlelength=1, handleheight=1)
# Set colorbar for link
gv.set_colorbar(fig, bar_colors=[normal_color, inverted_color], alpha=alpha, vmin=min_identity, bar_height=0.15, bar_label="Identity", bar_labelsize=10)
fig.savefig("example06.png", bbox_inches="tight")
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Hashes for pygenomeviz-0.0.9-py3-none-any.whl
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 | f506d003f3698e466ebf05cdc5acfe740cd7860f6993cd6de3e2c4352563278c |
|
| MD5 | 4235672039aeef460ed82d6e62762676 |
|
| BLAKE2b-256 | 2aaa8e93f3b73b7dff75cb2982bf3f0d244c30c2ba09699a8d003aee8d0464a0 |







