A genome visualization python package for comparative genomics

Project description
pyGenomeViz






[!NOTE] A major version upgrade, pyGenomeViz v1.0.0, was released on 2024/05. Backward incompatible changes have been made between v1.0.0 and v0.X.X to make for a more sophisticated API/CLI design. Therefore, v0.X.X users should pin the version to v0.4.4 or update existing code for v1.0.0. Previous v0.4.4 documentation is available here.
Table of contents
- Overview
- Installation
- API Examples
- CLI Examples
- GUI (Web Application)
- HTML Viewer
- Inspiration
- Circular Genome Visualization
- Star History
Overview
pyGenomeViz is a genome visualization python package for comparative genomics implemented based on matplotlib. This package is developed for the purpose of easily and beautifully plotting genomic features and sequence similarity comparison links between multiple genomes. It supports genome visualization of Genbank/GFF format file and can be saved figure in various formats (JPG/PNG/SVG/PDF/HTML). User can use pyGenomeViz for interactive genome visualization figure plotting on jupyter notebook, or automatic genome visualization figure plotting in genome analysis scripts/workflow.
For more information, please see full documentation here.
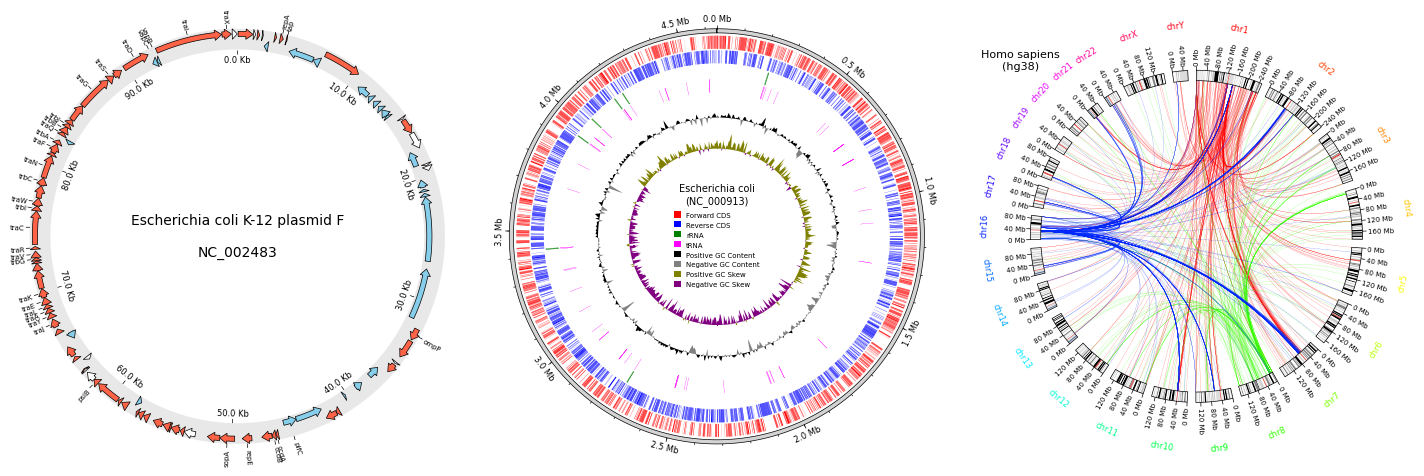
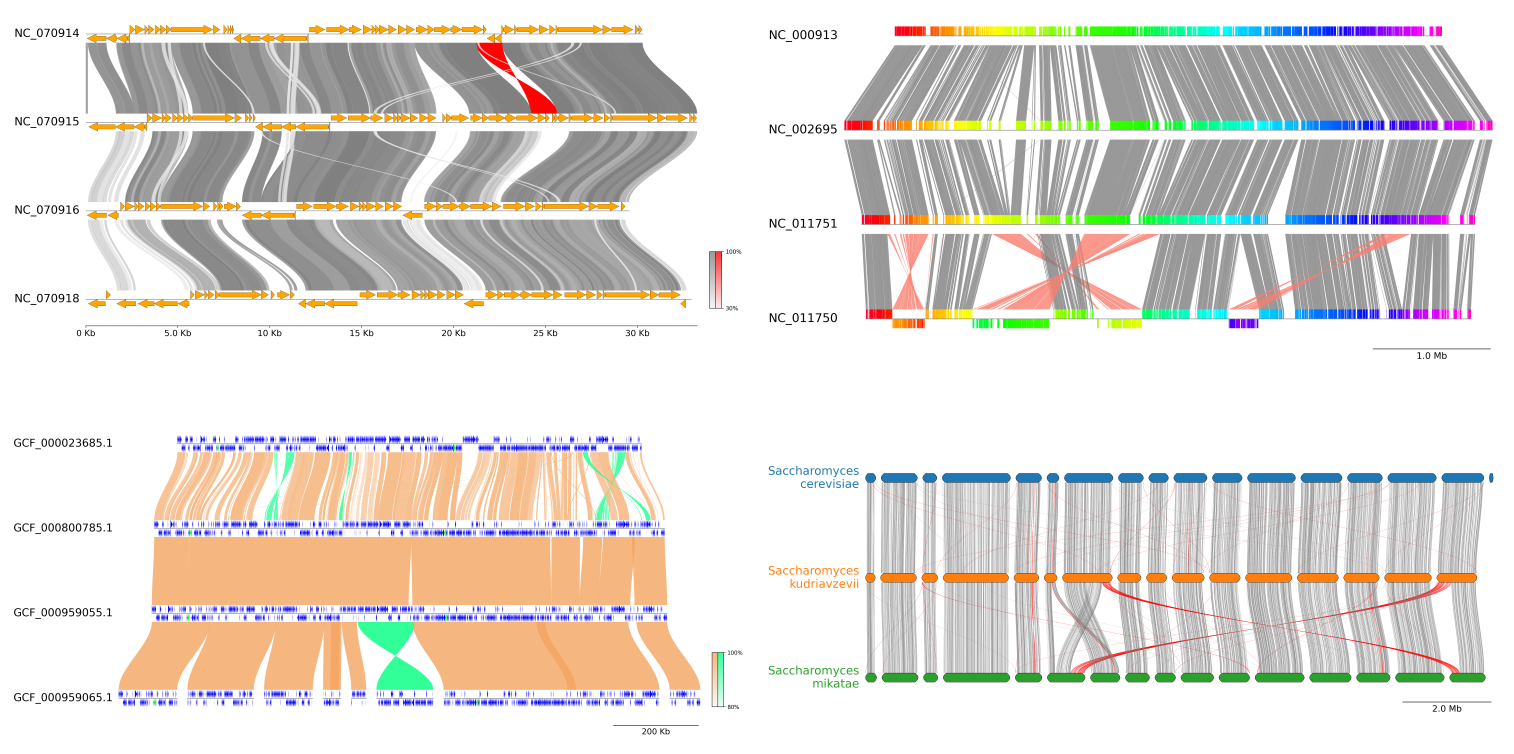
Fig.1 pyGenomeViz example plot gallery

Installation
Python 3.9 or later is required for installation.
Install PyPI package:
pip install pygenomeviz
Install conda-forge package:
conda install -c conda-forge pygenomeviz
Use Docker (Image Registry):
docker run -it --rm -p 8501:8501 ghcr.io/moshi4/pygenomeviz:latest pgv-gui -h
API Examples
Jupyter notebooks containing code examples below is available here.
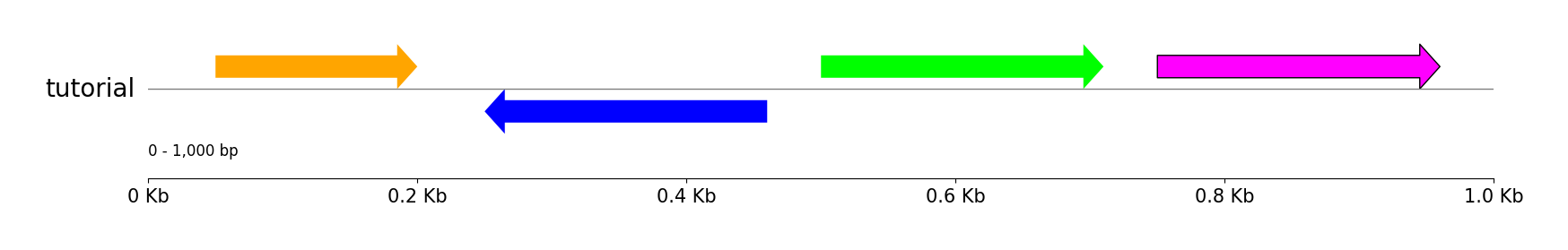
Features
from pygenomeviz import GenomeViz
gv = GenomeViz()
gv.set_scale_xticks(ymargin=0.5)
track = gv.add_feature_track("tutorial", 1000)
track.add_sublabel()
track.add_feature(50, 200, 1)
track.add_feature(250, 460, -1, fc="blue")
track.add_feature(500, 710, 1, fc="lime")
track.add_feature(750, 960, 1, fc="magenta", lw=1.0)
gv.savefig("features.png")
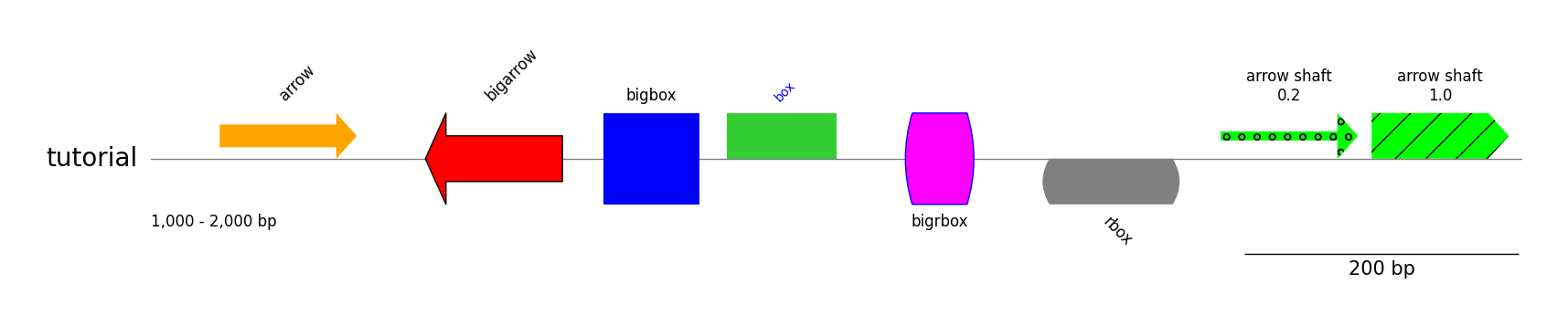
Styled Features
from pygenomeviz import GenomeViz
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
track = gv.add_feature_track("tutorial", (1000, 2000))
track.add_sublabel()
track.add_feature(1050, 1150, 1, label="arrow")
track.add_feature(1200, 1300, -1, plotstyle="bigarrow", label="bigarrow", fc="red", lw=1)
track.add_feature(1330, 1400, 1, plotstyle="bigbox", label="bigbox", fc="blue", text_kws=dict(rotation=0, hpos="center"))
track.add_feature(1420, 1500, 1, plotstyle="box", label="box", fc="limegreen", text_kws=dict(size=10, color="blue"))
track.add_feature(1550, 1600, 1, plotstyle="bigrbox", label="bigrbox", fc="magenta", ec="blue", lw=1, text_kws=dict(rotation=0, vpos="bottom", hpos="center"))
track.add_feature(1650, 1750, -1, plotstyle="rbox", label="rbox", fc="grey", text_kws=dict(rotation=-45, vpos="bottom"))
track.add_feature(1780, 1880, 1, fc="lime", hatch="o", arrow_shaft_ratio=0.2, label="arrow shaft\n0.2", text_kws=dict(rotation=0, hpos="center"))
track.add_feature(1890, 1990, 1, fc="lime", hatch="/", arrow_shaft_ratio=1.0, label="arrow shaft\n1.0", text_kws=dict(rotation=0, hpos="center"))
gv.savefig("styled_features.png")
Tracks & Links
from pygenomeviz import GenomeViz
genome_list = [
dict(name="genome 01", size=1000, features=((150, 300, 1), (500, 700, -1), (750, 950, 1))),
dict(name="genome 02", size=1300, features=((50, 200, 1), (350, 450, 1), (700, 900, -1), (950, 1150, -1))),
dict(name="genome 03", size=1200, features=((150, 300, 1), (350, 450, -1), (500, 700, -1), (700, 900, -1))),
]
gv = GenomeViz(track_align_type="center")
gv.set_scale_bar()
for genome in genome_list:
name, size, features = genome["name"], genome["size"], genome["features"]
track = gv.add_feature_track(name, size)
track.add_sublabel()
for idx, feature in enumerate(features, 1):
start, end, strand = feature
track.add_feature(start, end, strand, plotstyle="bigarrow", lw=1, label=f"gene{idx:02d}", text_kws=dict(rotation=0, vpos="top", hpos="center"))
# Add links between "genome 01" and "genome 02"
gv.add_link(("genome 01", 150, 300), ("genome 02", 50, 200))
gv.add_link(("genome 01", 700, 500), ("genome 02", 900, 700))
gv.add_link(("genome 01", 750, 950), ("genome 02", 1150, 950))
# Add links between "genome 02" and "genome 03"
gv.add_link(("genome 02", 50, 200), ("genome 03", 150, 300), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 350, 450), ("genome 03", 450, 350), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 02", 900, 700), ("genome 03", 700, 500), color="skyblue", inverted_color="lime", curve=True)
gv.add_link(("genome 03", 900, 700), ("genome 02", 1150, 950), color="skyblue", inverted_color="lime", curve=True)
gv.savefig("tracks_and_links.png")

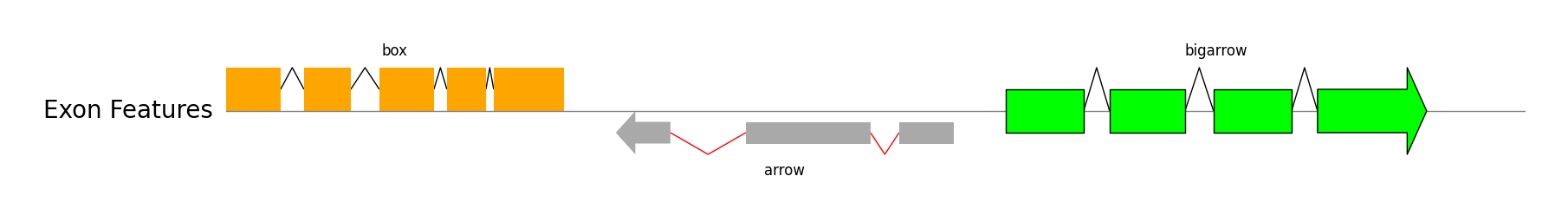
Exon Features
from pygenomeviz import GenomeViz
exon_regions1 = [(0, 210), (300, 480), (590, 800), (850, 1000), (1030, 1300)]
exon_regions2 = [(1500, 1710), (2000, 2480), (2590, 2800)]
exon_regions3 = [(3000, 3300), (3400, 3690), (3800, 4100), (4200, 4620)]
gv = GenomeViz()
track = gv.add_feature_track("Exon Features", 5000)
track.add_exon_feature(exon_regions1, strand=1, plotstyle="box", label="box", text_kws=dict(rotation=0, hpos="center"))
track.add_exon_feature(exon_regions2, strand=-1, plotstyle="arrow", label="arrow", text_kws=dict(rotation=0, vpos="bottom", hpos="center"), patch_kws=dict(fc="darkgrey"), intron_patch_kws=dict(ec="red"))
track.add_exon_feature(exon_regions3, strand=1, plotstyle="bigarrow", label="bigarrow", text_kws=dict(rotation=0, hpos="center"), patch_kws=dict(fc="lime", lw=1))
gv.savefig("exon_features.png")
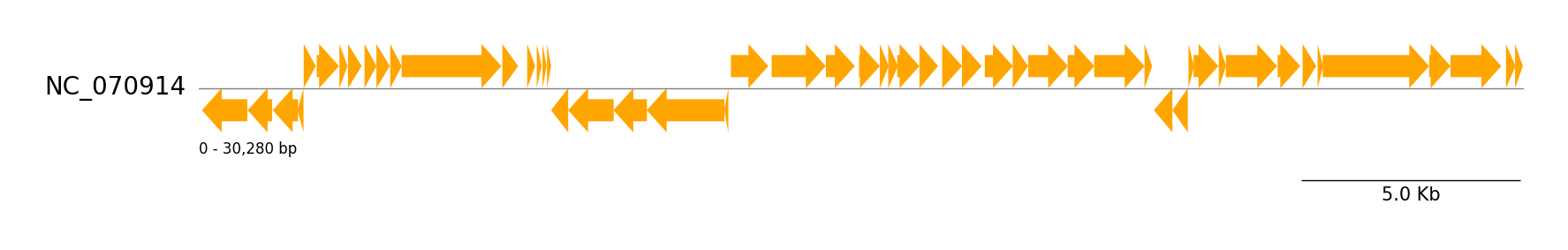
Genbank Features
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Genbank
from pygenomeviz.utils import load_example_genbank_dataset
gbk_files = load_example_genbank_dataset("yersinia_phage")
gbk = Genbank(gbk_files[0])
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
track = gv.add_feature_track(gbk.name, gbk.genome_length)
track.add_sublabel()
features = gbk.extract_features()
track.add_features(features)
gv.savefig("genbank_features.png")
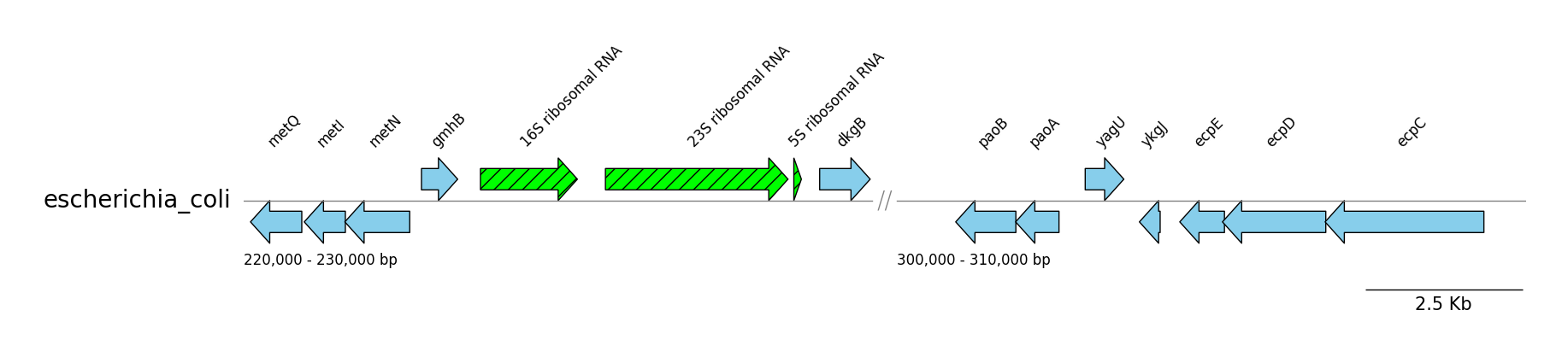
GFF Features
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Gff
from pygenomeviz.utils import load_example_gff_file
gff_file = load_example_gff_file("escherichia_coli.gff.gz")
gff = Gff(gff_file)
gv = GenomeViz()
gv.set_scale_bar(ymargin=0.5)
target_ranges = ((220000, 230000), (300000, 310000))
track = gv.add_feature_track(name=gff.name, segments=target_ranges)
track.set_segment_sep(symbol="//")
for segment in track.segments:
segment.add_sublabel()
# Plot CDS features
cds_features = gff.extract_features(feature_type="CDS", target_range=segment.range)
segment.add_features(cds_features, label_type="gene", fc="skyblue", lw=1.0)
# Plot rRNA features
rrna_features = gff.extract_features(feature_type="rRNA", target_range=segment.range)
segment.add_features(rrna_features, label_type="product", hatch="//", fc="lime", lw=1.0)
gv.savefig("gff_features.png")
GFF Contigs
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Gff
from pygenomeviz.utils import load_example_gff_file, is_pseudo_feature
gff_file = load_example_gff_file("mycoplasma_mycoides.gff")
gff = Gff(gff_file)
gv = GenomeViz(fig_track_height=0.5, feature_track_ratio=0.5)
gv.set_scale_xticks(labelsize=10)
# Plot CDS, rRNA features for each contig to tracks
for seqid, size in gff.get_seqid2size().items():
track = gv.add_feature_track(seqid, size, labelsize=15)
track.add_sublabel(size=10, color="grey")
cds_features = gff.get_seqid2features(feature_type="CDS")[seqid]
# CDS: blue, CDS(pseudo): grey
for cds_feature in cds_features:
color = "grey" if is_pseudo_feature(cds_feature) else "blue"
track.add_features(cds_feature, color=color)
# rRNA: lime
rrna_features = gff.get_seqid2features(feature_type="rRNA")[seqid]
track.add_features(rrna_features, color="lime")
gv.savefig("gff_contigs.png")

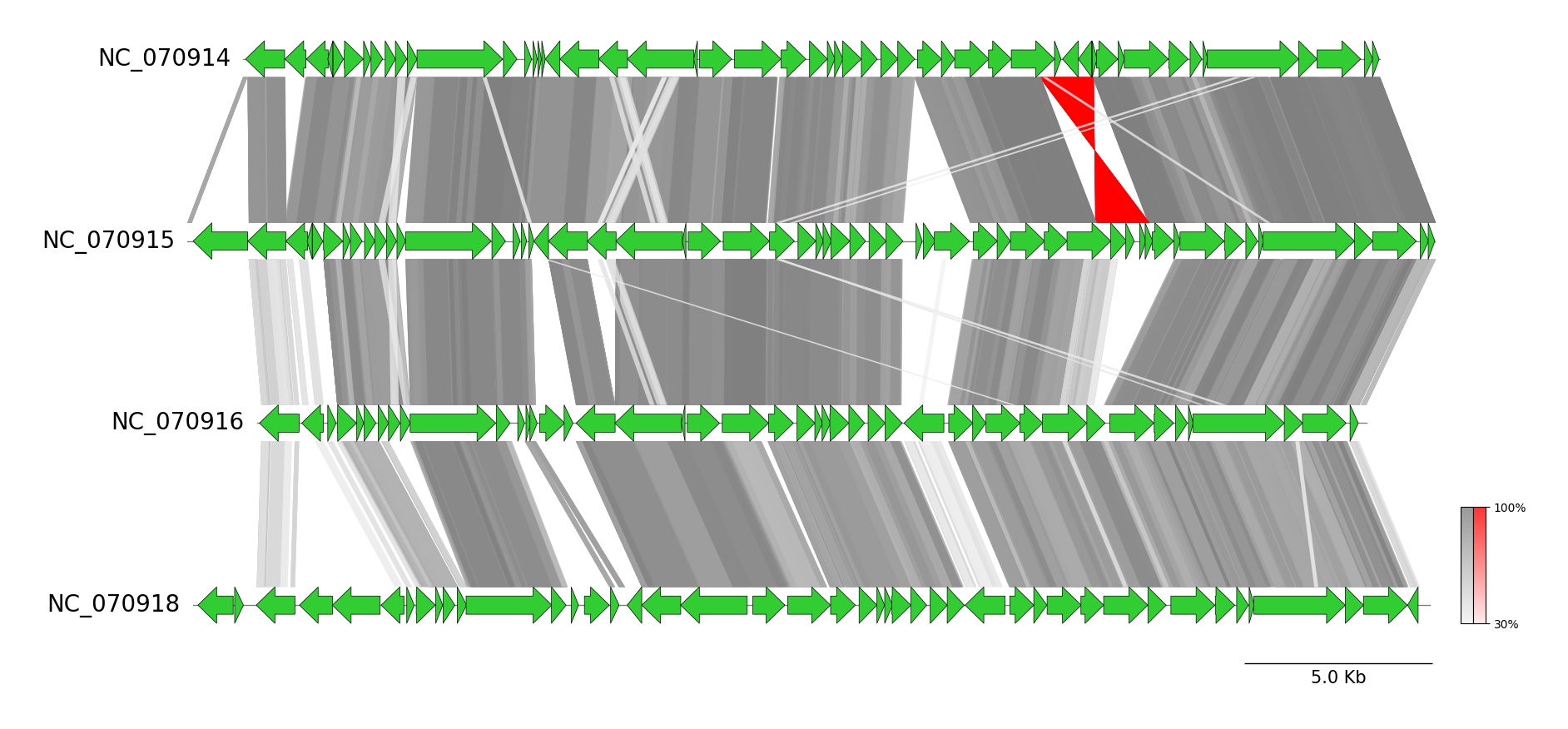
Genbank Comparison by BLAST
from pygenomeviz import GenomeViz
from pygenomeviz.parser import Genbank
from pygenomeviz.utils import load_example_genbank_dataset
from pygenomeviz.align import Blast, AlignCoord
gbk_files = load_example_genbank_dataset("yersinia_phage")
gbk_list = list(map(Genbank, gbk_files))
gv = GenomeViz(track_align_type="center")
gv.set_scale_bar()
# Plot CDS features
for gbk in gbk_list:
track = gv.add_feature_track(gbk.name, gbk.get_seqid2size(), align_label=False)
for seqid, features in gbk.get_seqid2features("CDS").items():
segment = track.get_segment(seqid)
segment.add_features(features, plotstyle="bigarrow", fc="limegreen", lw=0.5)
# Run BLAST alignment & filter by user-defined threshold
align_coords = Blast(gbk_list, seqtype="protein").run()
align_coords = AlignCoord.filter(align_coords, length_thr=100, identity_thr=30)
# Plot BLAST alignment links
if len(align_coords) > 0:
min_ident = int(min([ac.identity for ac in align_coords if ac.identity]))
color, inverted_color = "grey", "red"
for ac in align_coords:
gv.add_link(ac.query_link, ac.ref_link, color=color, inverted_color=inverted_color, v=ac.identity, vmin=min_ident)
gv.set_colorbar([color, inverted_color], vmin=min_ident)
gv.savefig("genbank_comparison_by_blast.png")
CLI Examples
pyGenomeViz provides CLI workflows for genome alignment result visualization of
Genbank genomes using BLAST / MUMmer / MMseqs / progressiveMauve, respectively.
BLAST CLI Workflow
See pgv-blast document for details.
# Download example dataset
pgv-download yersinia_phage
# Run BLAST CLI workflow
pgv-blast NC_070914.gbk NC_070915.gbk NC_070916.gbk NC_070918.gbk \
-o pgv-blast_example --seqtype protein --show_scale_bar --curve \
--feature_linewidth 0.3 --length_thr 100 --identity_thr 30
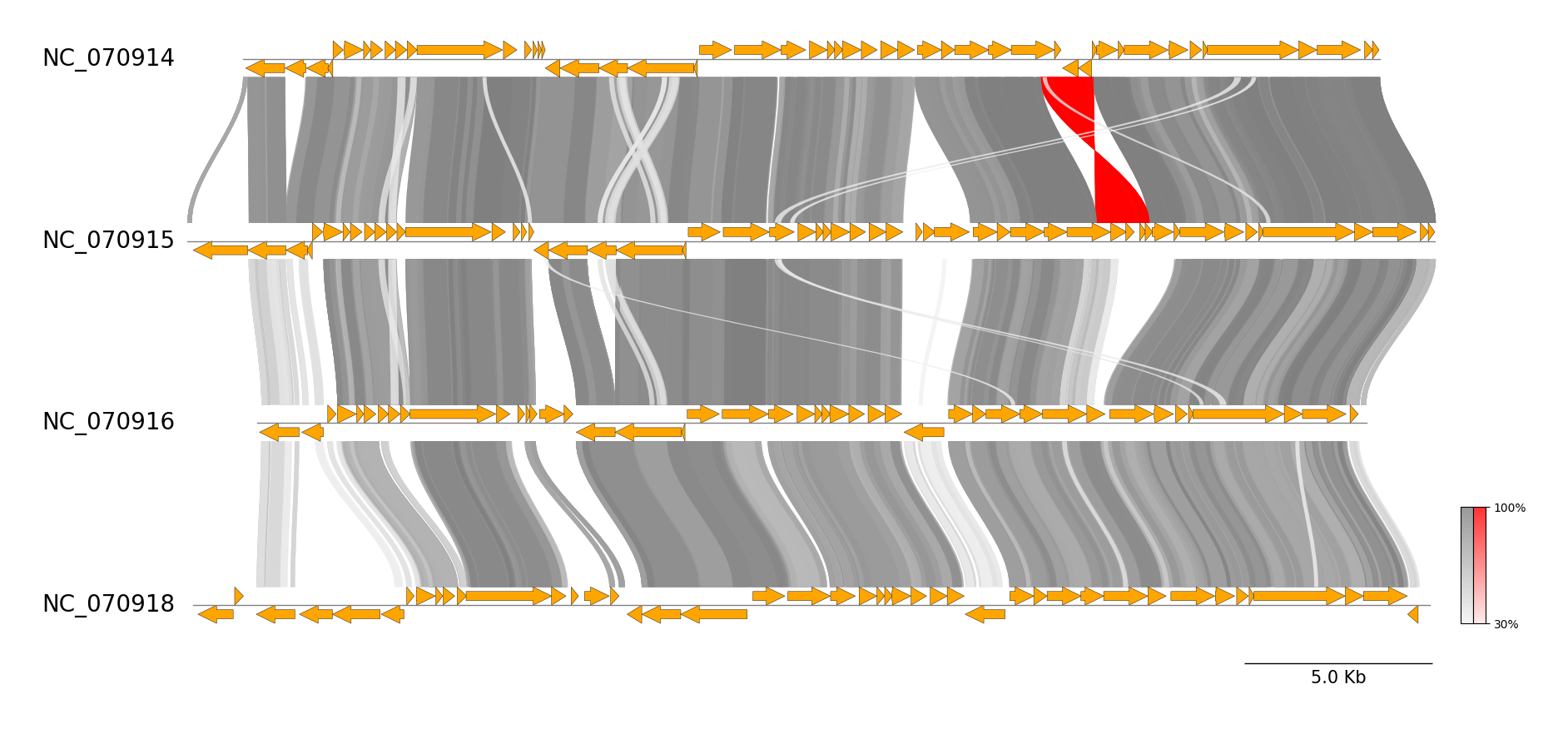
MUMmer CLI Workflow
See pgv-mummer document for details.
# Download example dataset
pgv-download mycoplasma_mycoides
# Run MUMmer CLI workflow
pgv-mummer GCF_000023685.1.gbff GCF_000800785.1.gbff GCF_000959055.1.gbff GCF_000959065.1.gbff \
-o pgv-mummer_example --show_scale_bar --curve \
--feature_type2color CDS:blue rRNA:lime tRNA:magenta

MMseqs CLI Workflow
See pgv-mmseqs document for details.
# Download example dataset
pgv-download enterobacteria_phage
# Run MMseqs CLI workflow
pgv-mmseqs NC_013600.gbk NC_016566.gbk NC_019724.gbk NC_024783.gbk NC_028901.gbk NC_031081.gbk \
-o pgv-mmseqs_example --show_scale_bar --curve --feature_linewidth 0.3 \
--feature_type2color CDS:skyblue --normal_link_color chocolate --inverted_link_color limegreen
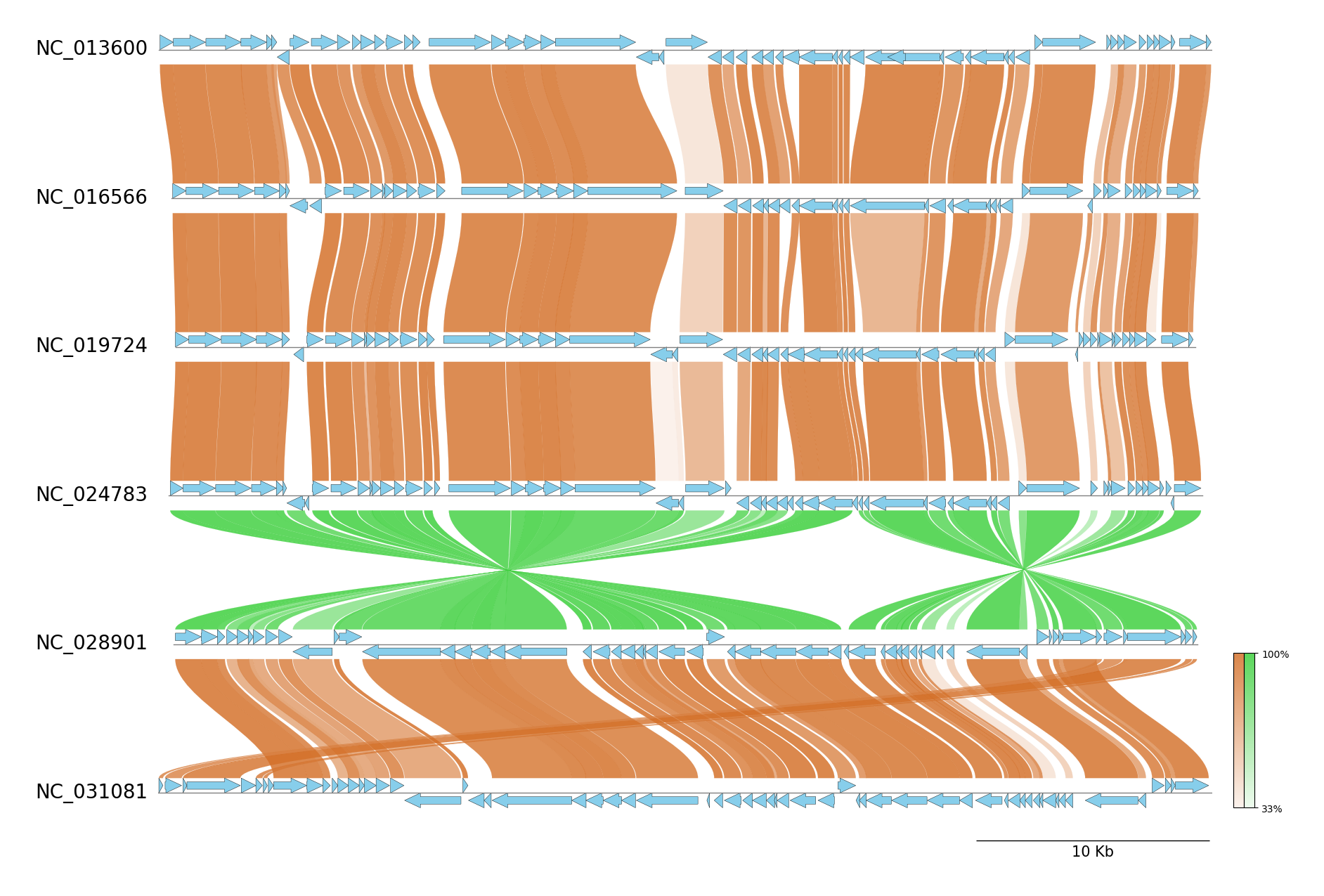
progressiveMauve CLI Workflow
See pgv-pmauve document for details.
# Download example dataset
pgv-download escherichia_coli
# Run progressiveMauve CLI workflow
pgv-pmauve NC_000913.gbk.gz NC_002695.gbk.gz NC_011751.gbk.gz NC_011750.gbk.gz \
-o pgv-pmauve_example --show_scale_bar

GUI (Web Application)
pyGenomeViz implements GUI (Web Application) functionality using streamlit as an option. Users can easily visualize the genomic features in Genbank files and their comparison results with GUI (Demo Page). See pgv-gui document for details.
HTML Viewer
pyGenomeViz implements HTML viewer output functionality for interactive data visualization.
In API, HTML file can be output using savefig_html method. In CLI, user can select HTML file output option.
As shown below, pan/zoom, tooltip display, object color change, text change, etc are available in HTML viewer
(Demo Page1, Demo Page2).

Following libraries were used to implement HTML viewer.
- Spectrum: Colorpicker
- Panzoom: SVG panning and zooming
- Tabulator: Interactive Table
- Micromodal: Modal dialog
- Tippy.js: Tooltip
Inspiration
pyGenomeViz was inspired by
Circular Genome Visualization
pyGenomeViz is a python package designed for linear genome visualization. If you are interested in circular genome visualization, check out my other python package pyCirclize.
Fig. pyCirclize example plot gallery